Mechanism • Catalyst Design • Reaction Development • Complex Molecule Synthesis/Modification
OUR GENERAL APPROACH
We are chemists who consider catalysis and synthesis as our core specialty. We view ourselves as molecular designers keen on learning more about catalysis and how it might be used to generate functional molecules of all sizes. Our approach is multidisciplinary and collaborative.
Catalytic transformations that deliver stereochemically defined fragments, necessary to the function of a molecule, regardless of its size, is one central tenet of our research. For instance, we have developed catalysts that can be used to prepare a range of otherwise difficult-to-access E– or Z-trisubstituted alkenes in high stereoisomeric purity. Another example are catalytic strategies for forming C–prenyl bonds. We have taken on the task of designing catalysts that can be used to synthesize readily alterable tetrasubstituted olefins in either stereoisomeric form – processes that are of value to drug discovery and development. We introduce new multicomponent transformations, involving one or perhaps more catalysts, capable of delivering products that are densely functionalized and are easy to modify. Mechanistic understanding is central to our studies, and with each advance we hope to enrich this foundation.
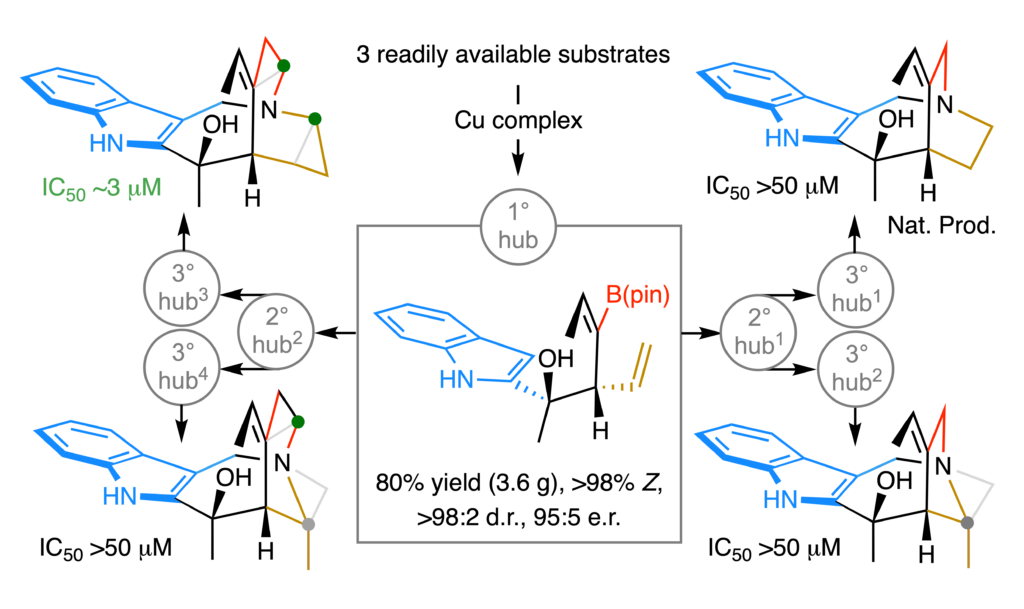
Our search is not only for a practical way of synthesizing natural products, it is also to develop strategies for securing, through networks of shared transformations, precisely re-modelled skeletal analogs. We recently put forward a catalytic multicomponent diastereo- and enantioselective process that generates a multi-functional and chemoselectively modifiable platform that can be used for concise synthesis of a rare polycyclic indole alkaloid. What is more, it facilitates access to several scaffolds that have been expanded, contracted or distorted by one-two methylene units. Together with the Hergenrother group (UIUC) we established that a doubly-expanded skeletal analog of the aforementioned weakly anti-malarial natural product – but not itself or the other exactly altered frameworks – is cytotoxic against the four cancer cell lines screened (3 mM). Molecular dynamics and AI-accelerated docking studies performed by the Liu group (Pittsburgh) and Accutar Biotech (Shanghai) helped us gain insight regarding what differentiates various scaffolds and what might be the origins of their marked proclivity for binding to various biological receptors. On an associated front, we are keen on developing efficient ways of surgically altering scaffolds of complex bioactive molecules; this would facilitate drug discovery by making it possible to explore rarely visited regions in the diversity space.
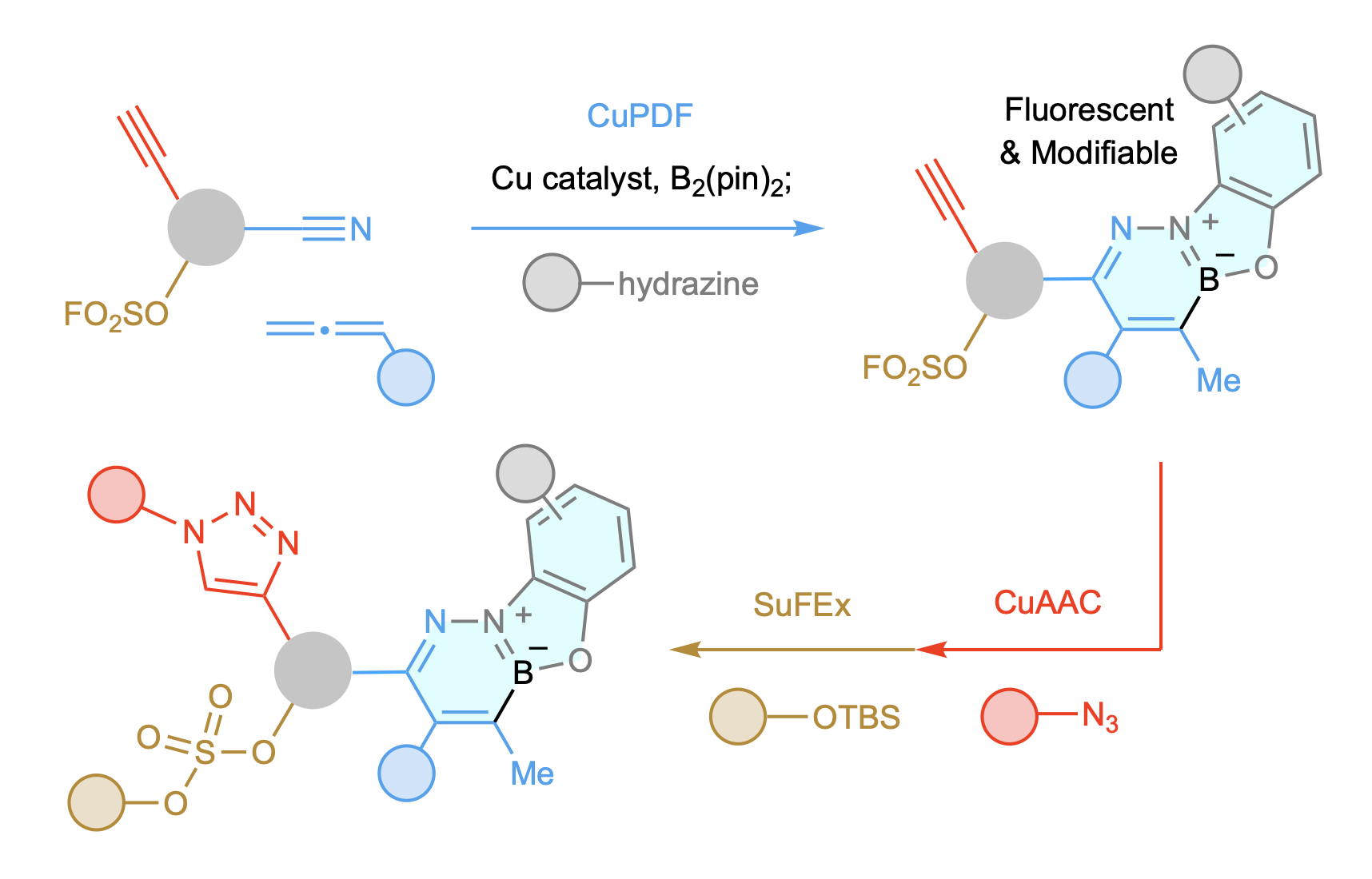
We favor investigating catalytic approaches that can expedite access to new molecules, small (bioactive) and large (functional polymers). We have had great fun designing a link-and-modify strategy that is orthogonal to CuAAC and SuFEx. By merging these three catalytic processes, one can quickly fabricate site-selectively modifiable and/or cleavable macromolecules. The strategy can be used for concise synthesis of multi-drug conjugates that also have fluorescent linkages.
What we will be searching for in five to ten years we do not know. What we can tell you is that, if you join us, you will have something to say about it.
RECENT PUBLICATIONS
December 13, 2023
Nature Chemistry
“Click Processes Orthogonal to CuAAC and SuFEx Forge Selectively Modifiable Fluorescent Linkers“
The appeal of catalytic click chemistry is owed to copper-catalyzed azide-alkyne cycloaddition (CuAAC), a process that is orthogonal to the more recently introduced sulfur-fluoride exchange (SuFEx). However, the triazole rings generated by CuAAC are not readily modifiable and SuFEx connectors cannot be selectively functionalized, attributes that would be attractive in a click process. We have been able to develop a link and in situ modify strategy for merging a nitrile, an allene, a diborane, and a hydrazine. We refer to this process as bisphosphine–copper-catalyzed phenoxydiazaborinine formation, or CuPDF. In addition, we have developed a version of the process that can be performed in aqueous media. This variant involves the use of an aniline as the modifier, and we call it copper- and palladium-catalyzed quinoline formation or Cu/PdQNF. CuPDF and Cu/PdQNF are easy to perform and deliver robust, alterable, and tunable fluorescent hubs. CuPDF and Cu/PdQNF are orthogonal to SuFEx and CuAAC, despite the latter and CuPDF being also catalysed by an organocopper species. We have applied these methods to protecting group-free syntheses of sequence-defined branched oligomers, a chemoselectively amendable polymer, three drug conjugates, and a two-drug conjugate. Development of new click processes that generate uniquely functional linkers while being orthogonal to other key catalytic reactions is an exciting new direction for our group.
February 19, 2024
Nature Chemistry
“A Catalytic Process Enables Efficient and Programmable Access to Precisely Altered Indole Alkaloid Scaffolds“
A compound’s contour directly impacts its ability to elicit biological response, rendering access to distinctly shaped molecules desirable. Modification of a natural product’s framework is a common tactic, but possible only if the material is abundant and contains suitably modifiable functional groups. As part of a new program in our laboratories, we have developed a programmable strategy for concise synthesis of precisely altered bridged polycyclic alkaloid scaffolds. The approach is supported by a scalable catalytic diastereo- and enantioselective multicomponent process that delivers tertiary homoallylic alcohols bearing chemo- and regioselectively differentiable alkenyl moieties. One of the products was used to launch concise and progressively divergent syntheses of a naturally occurring indole alkaloid and precisely expanded, contracted, and/or distorted frameworks (average number of steps/scaffold = 7). In vitro testing shows that a skeleton by one methylene expanded in two regions is cytotoxic against four types of cancer cell lines. Mechanistic and density functional theory (DFT) studies provide rationales for several unanticipated selectivity trends.